
TEA y Síndrome de Martin-Bell
¿Qué es?
En 1943, Martin y Bell descubrieron un tipo de deficiencia mental hereditario ligado al cromosoma X, que hoy conocemos como síndrome del X frágil.
n 1969, Lubs estudió una familia en la que cuatro varones de tres generaciones diferentes presentaban deficiencia mental. Los estudios citogenéticos de las muestras de estos pacientes revelaron una constricción inusual en el brazo largo del cromosoma X en el 10-33% de las células en cultivo.
En un estudio posterior de la misma familia, Lubs y col., en 1984, describieron rasgos faciales inusuales en los miembros de esta familia que presentaban la afección: caras alargadas, orejas largas con inserción más baja de lo habitual, rasgos faciales asimétricos y cejas prominentes.
También en 1969, Opitz y col. emplearon el término “síndrome de Martin-Bell” para referirse a un caso de deficiencia mental familiar con características de dicho síndrome. En aquel entonces, nadie había relacionado el síndrome de Martin-Bell con el síndrome del X frágil de Lub.
En 1981, Richards y col. demostraron que ambos síndromes eran en realidad el mismo trastorno. Para ello, estudiaron a la misma familia que habían descrito Martin y Bell y utilizando la técnica de cultivo empleada por Lubs, observaron que todos los varones afectados presentaban el sitio frágil del cromosoma X en el 5-17% de sus células en cultivo.
En 1991, Verkerk y col. describieron un gen asociado al trastorno: el gen FMR-1 (acrónimo inglés de Fragile X linked Mental Retardation type 1; deficiencia mental ligado al X de tipo 1). Este descubrimiento ha traído consigo grandes mejoras en el diagnóstico prenatal y en la identificación de personas afectadas y en el rango de premutación.
El Síndrome de Martin- Bell o X-frágil es un trastorno genético y es la forma más común de discapacidad intelectual hereditaria,que causa síntomas similares al TEA.
El nombre se refiere a una parte del cromosoma X que tiene una porción defectuosa que, al observarse a través del microscopio, aparece comprimida y frágil.
Está causada por la ausencia de una proteína llamada proteína de retraso mental frágil (FMRP, por sus siglas en inglés). La FMRP está implicada tanto en el desarrollo cognitivo como en la función reproductora de la mujer.
Los personas con este síndrome carecen de la proteína FMR debido a una mutación en el gen que codifica su síntesis, el FMR1. Se encuentra en el cromosoma X y la alteración concreta que causa la anomalía es una repetición de una secuencia concreta del gen: el trinucleótido CGG.
El número de repeticiones de esta secuencia determina que los individuos sean:
Sanos: el número de repeticiones oscila entre 6 y 54.
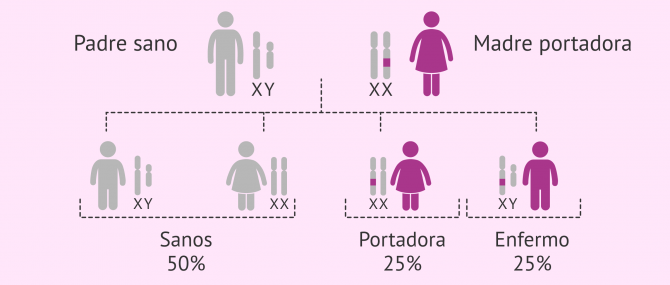
Portadores: tienen entre 55 y 200 repeticiones (premutación) y transmiten el síndrome aunque no lo padezcan.
Afectados: presentan más de 200 repeticiones, tienen la mutación completa y, por tanto, la enfermedad.
El síndrome X frágil resulta de un cambio, llamado mutación, de un gen único.
¿Cuál es su prevalencia?
Aproximadamente 1 de cada 3 niños que tienen el síndrome X frágil también cumplen con los criterios para el diagnóstico del TEA y cerca de 1 de cada 25 niños diagnosticados con el TEA presentan la mutación que causa el síndrome X frágil.
Debido a que este trastorno se hereda, se debe examinar a los niños con el TEA en busca de X frágil, especialmente si los padres desean tener más hijos. Otros miembros de la familia que estén planeando tener hijos también pueden querer examinarse en busca del síndrome X frágil.
Fenotipo y síntomas:
Las características de este síndrome son variadas, pero la más común es el retraso mental, que puede ser desde leve a grave. También existen algunos rasgos físicos frecuentes:
- Orejas grandes y salidas.
- Cara alargada y estrecha.
- Mandíbula inferior prominente.
- Deficiencias auditivas y visuales.
- Hiperlaxitud (mayor flexibilidad) en las articulaciones y tono muscular bajo.
- Cuerpo grande.
- Piel suave.
- Problemas cardíacos y epilepsia.
- Macroorquidismo (testículos más grandes de lo habitual) en la adolescencia.
- Pies planos.
- Retraso psicomotor.
- Hiperactividad, impulsividad y falta de atención.
- Trastornos del lenguaje, la lectura y la escritura.
- Rasgos similares a personas con TEA (timidez, aleteos con las manos, lenguaje repetitivo…).
Métodos diagnósticos
El diagnóstico no puede basarse sólo en el cuadro clínico, ya que los rasgos físicos pueden ser leves o estar ausentes, de modo que se debe confirmar con un test genético del gen FMR1, que se debe realizar en todos los pacientes con una deficiencia intelectual o con autismo.
El diagnóstico diferencial incluye otras deficiencias intelectuales ligadas al cromosoma X, el síndrome de Sotos, los síndromes asociados a microdeleciones de material genético (como el síndrome de la deleción 22q11.2), el síndrome de alcoholismo fetal (véanse estos términos) y los trastornos del espectro autista de origen idiopático.






